Designing Media for the Cancer
Leverage the 20+ years of cancer cell culturing knowledge imbedded in our cancer cell culturing media. Saves you time and will rescue your research and your business. Promise.

Available Media for Cancer Cells
Leverage the awesome cancer cell culturing media that saves you time and will rescue your discovery research. Promise.

Partnering
Leverage the combination of cancer cell technologies and drug discovery for your research projects. Partnering doesn't have to be difficult and it can save money. Promise.
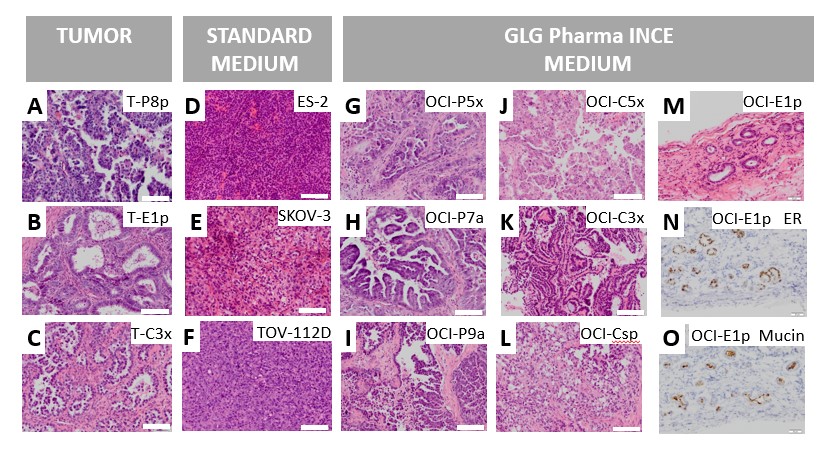
An Example of Our Media for Cancer Cell Culturing
Epithelial Example: Ovarian Tumor Cells
In xenographs, morphology is also better preserved